Abstract
The patient, D.P., was a Caucasian male who was diagnosed in 1989 with a null-cell or nonfunctioning pituitary adenoma at age 29. His only symptoms at that time were visual field changes and fatigue. He was seen by an ophthalmologist for his complaints of loss of acuity and diplopia. A subsequent MRI of the brain revealed a pituitary mass involving the optic chiasm. He was treated with surgical ablation through the trans-sphenoidal approach followed by adjuvant stereotactic radiosurgery. Postsurgically, he received 43 Gy of radiation with a tumor margin dose of 20 Gy. Due to his treatment, the patient became dependent on hormonal replacement; testosterone (200 mg IM every 3 weeks), thyroid (levothyroxine 125 ìg daily), and hydrocortisone (20 mg in the morning and afternoon and 10 mg at bedtime) were necessary. His hormone levels were monitored by his primary care physician, who adjusted dosages as needed.
D.P. was followed closely without evidence of recurrence until 1994, when he was found to have developed an enhancing mass in the middle and infratemporal cranial fossa. A complete resection was performed; the pathology was positive for radiation-induced meningioma with bone involvement.
Further resection for recurrent tumor was performed in 1998 and 2000. Following the resection in 2000, D.P. had right-sided weakness and eventual hemiplegia. Additionally, he had a complete absence of third and sixth nerve function and complete blindness in his left eye (Figure 2). In 2004, he experienced a recurrence, now with a left-sided middle fossa and intraorbital meningioma and involvement of the ocular contents.

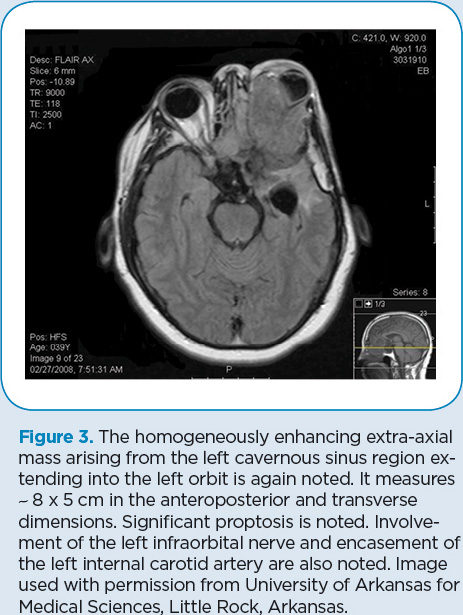
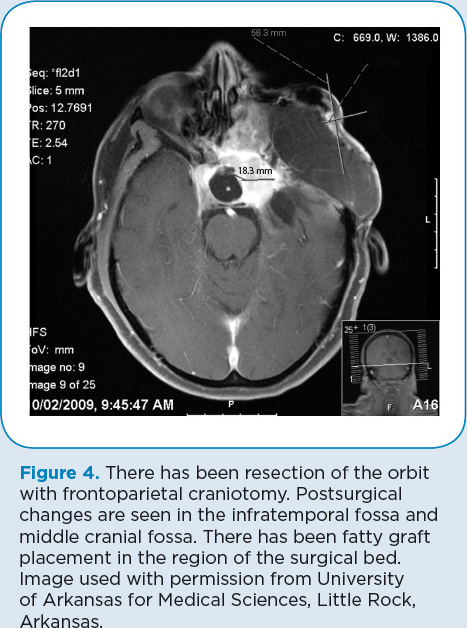
In April 2008 (Figure 3), D.P. was found to have a massive recurrence of his tumor that involved the paranasal sinuses, bone, and soft tissues as well as the peripapillary choroid intraocularly and the epibulbar long and short posterior ciliary arteries and nerves. Resection was done, including complete enucleation of the left eye. Additional reconstruction of the skull was performed during a 20-hour surgical procedure (Figure 4).
Medical Treatment Record
In November 2003, D.P. began palliative treatment with chemotherapy. (See Table 1 for a summary of D.P.’s medical treatment throughout his disease course.) He was initially treated with interferon-alpha, 1 million units SC daily, augmented with tamoxifen 10 mg twice daily (Sioka & Kyritsis, 2009; Rockhill, Mrugala, & Chamberlain, 2007). He tolerated the treatment with few problems. This regimen was stopped in August 2004, when his disease was found to have progressed. Incomplete surgical resection was performed, and D.P. was started on targeted therapy with gefitinib (Iressa) 250 mg daily (Norden, Drappatz, & Wen, 2007). This was continued until April 2005, when he was again found to have disease progression on MRI. Rather than repeat resection at this time, the patient chose to change medical treatment in the hopes of stopping or shrinking his tumor.

In July 2005, D.P.’s treatment was changed to single-agent bevacizumab (Avastin) 10 mg/kg IV every 14 days (Norden et al., 2007). Initially, a reduction in tumor size was noted, but after 2 months, the agent was discontinued due to tumor growth. During treatment with bevacizumab, the patient experienced hypertension and required treatment with triamterene/hydrochlorothiazide (37.5 mg/25 mg) daily. Upon discontinuing the bevacizumab, the treatment was changed to hydroxyurea 500 mg orally twice daily (Newton, 2007). D.P. was able to tolerate this treatment with few side effects or complications. He continued this drug with relative stability of his tumor until December 2006, at which time he had further progression on MRI so he was referred back to neurosurgery.
After consulting with the neurosurgery team, D.P. again decided to try changing his medical treatment rather than opt for a repeat resection. He was started on sorafenib (Nexavar) 400 mg twice daily (Newton, 2007). He was unable to tolerate the full dose due to grade 3 diarrhea, palmar-plantar erythrodysesthesia, and grade 2 neutropenia. The dose was lowered to 200 mg twice daily, which was further reduced after 2 weeks to an every-other-day schedule due to continuing problems. Eventually, D.P. was taken off sorafenib due to the toxicities described above. In July 2007, he was started on sunitinib (Sutent; Newton, 2007) and experienced fewer side effects. He was maintained on a dose of 50 mg daily until March 2008, at which time his tumor had progressed to a substantial size, requiring repeat resection.
Following the April 2008 resection, D.P. again started on bevacizumab with the addition of liposomal doxorubicin (Doxil; Travitzky, Libson, Nemirovsky, Hadas, & Gabizon, 2003). Due to logistic constraints, past history with hypertension, and dosing schedules, it was decided to treat the patient with monthly, rather than the customary twice monthly, bevacizumab, and monthly liposomal doxorubicin. The bevacizumab was dosed at 15 mg/kg IV, followed by the liposomal doxorubicin at 30 mg/m² IV. He was maintained on this regimen with stable tumor size for 24 months, with no apparent adverse effects.
In January 2011, D.P. was seen in the hospital for weakness, fatigue, and neurologic changes. An MRI revealed a thromboembolic event in his brain, possibly associated with the use of bevacizumab. At that time, bevacizumab was stopped and the patient was discharged home on hospice. He expired 2 days after returning home, at the age of 42.